|
| |
|
Revista
Electrónica de Medicina Intensiva
Artículo nº C23. Vol 4 nº 12, diciembre 2004.
Autor: Vicente Gómez Tello
|
[ Anterior ] [ Arriba ] [ Siguiente ]
|
|
El hígado es un órgano central en la
homeostasis, con importantes funciones metabólicas, inmunológicas, y
depurativas de sustancias tóxicas endógenas y exógenas. Desde tiempos de
Hipócrates se sabe que estas funciones pueden alterarse precozmente por
fenómenos asociados a la sepsis, tales como hipermetabolismo, hipotensión
e isquemia tisular. Así, y de manera secundaria, el hígado puede
convertirse en un órgano causante y perpetuador de fracaso multiorgánico.
|
1. Definición e incidencia |
La
disfunción hepática puede definirse de muchas maneras. Quizá la más común
sea la elevación de la bilirrubina por encima de 2 mg/dl, de acuerdo a los
criterios de la escala SOFA de fracaso multiorgánico [1]. A este criterio
puede asociarse elevación del INR, e incremento del 100% de los niveles de
transaminasas. No obstante, y como veremos, estos signos pueden ser
inespecíficos y tardíos con respecto al verdadero inicio de la afectación
hepática.
La
disfunción hepática en la sepsis no es infrecuente, estimándose su
incidencia en un 62% de enfermos sépticos ingresados en UCI [2] y en un
0,15% del total de enfermos críticos [3]. Sin embargo, si se excluyen
pacientes con hepatopatía o neoplasia, otros autores hablan de un 6 % [4].
El origen principal de la sepsis es el foco abdominal [5], y el agente más
frecuente E. coli.
La contribución de la disfunción
hepática a la mortalidad en la sepsis ha sido estudiada por Russell y col.
en una cohorte de 287 pacientes sépticos en UCI. La disfunción hepática
mantiene un coeficiente de mortalidad en un modelo de regresión de Cox de
1,3, por debajo de la disfunción vascular y neurológica, y similar a la
renal [6]. De ahí, el interés de un adecuado diagnóstico y tratamiento de
soporte.
El hígado
puede afectarse por la sepsis en dos momentos cronológicos, generando un
daño hepático primario y otro secundario con consecuencias patológicas
diferentes [7].
El daño
primario obedece a la disfunción orgánica vascular en el inicio de la
sepsis. La sepsis grave y el shock séptico pueden provocar hepatitis
isquémica por hipoperfusión. El hígado es un órgano relativamente
resistente a la isquemia, puesto que se precisa un 70% de reducción del
flujo sanguíneo para comprometer su extracción de oxígeno. Cuando esta
reserva se agota, la isquemia hepática produce depleción de ATP
mitocondrial y necrosis celular centrolobulillar. En función de la masa
celular afectada aparecen diferentes fenómenos analíticos y clínicos:
aumento de transaminasas de hasta 20 veces, hipoglucemia, alteración de
conciencia, acidosis láctica, coagulación intravascular diseminada (CID) e
insuficiencia renal [8]. Seeto y col. [9] realizaron un estudio
comparativo entre 31 enfermos que presentaron hipotensión mantenida
(tensión arterial media menor de 75 mm Hg durante más de 15 minutos) y
signos de hepatitis isquémica, frente a pacientes traumatizados con
hipotensión pero sin hepatitis. Los resultados mostraron que la hepatitis
se relacionaba en un 97% de los casos con la presencia de fracaso cardiaco
derecho por lesión subyacente. Concluyen que el fenómeno de la hepatitis
isquémica pudiera estar relacionado con un obstáculo al flujo a nivel
portal, o por daño crónico de los hepatocitos sometidos a disfunción
cardiaca derecha. Este hecho indica que el principal factor de la lesión
hepática isquémica puede no ser la hipoperfusión (shock, trauma, etc.),
necesitando de otros factores. De hecho, sólo 2 pacientes de 31
presentaron signos de sepsis, con lo que puede concluirse que la isquemia
no es el mecanismo predominante de daño hepático en la sepsis.
El daño secundario viene producido
por la acción de bacterias y endotoxinas. Esta disfunción celular parece
anterior e independiente a los fenómenos hemodinámicos primarios de la
sepsis, como se ha demostrado en modelos animales: en ratas, el flujo
hepático aumentado no parece prevenir la disfunción hepatocelular [10,
11]. La fisiopatología de esta lesión se explica en el siguiente apartado.
|
3. Fisiopatología e histología |
La
endotoxina bacteriana se une al macrófago hepático, o célula de Kupffer,
promoviendo la liberación de citokinas como el TNF-α y las interleuquinas
IL-1 e IL-6. Son estas moléculas quienes ejercen acciones a nivel
bioquímico y celular. A nivel bioquímico sobre la captación y transporte
de compuestos biliares, y a nivel celular mediante la interacción de
diversas células con daño final a través de los neutrófilos [8]. Podemos
distinguir dos niveles de lesión: el bioquímico y el celular.
3.1
A nivel bioquímico
La
formación de bilis es un proceso osmótico que comprende secreción activa
de solutos orgánicos e inorgánicos, más un transporte pasivo de agua. El
paso limitante en la formación de bilis es la captación de bilirrubina y
ácidos biliares a nivel de la porción sinusoidal (vascular) de los
hepatocitos. A nivel bioquímico la producción de citokinas origina un
transporte alterado de la bilirrubina y ácidos biliares conjugados en las
porciones basal y apical de la membrana celular [12]. La causa estriba en
un déficit de síntesis de un grupo de proteínas transportadoras de sales
biliares, y dependientes de los canales del sodio, conocidas como NTCP
[13, 14]. Por otro lado la secreción canalicular tiene lugar
principalmente por la acción de la proteína BSEP (bomba secretora de sales
biliares) dependiente de ATP [15]; otros sistemas ATP independientes
(MRP2, ABCC2, cMOAT) pueden transportar múltiples aniones orgánicos y
drogas [16] (Figura 1)
Figura 1
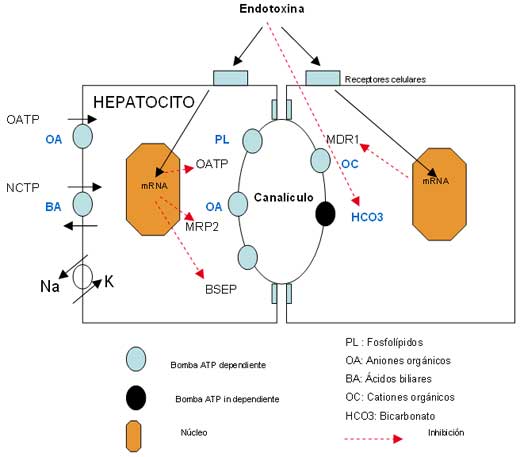
La
deficiencia de síntesis de estas proteínas se origina por la acción de la
endotoxina y citokinas a través de receptores de membrana (RAR), y
nucleares como el NFκB y el sistema HNF [12]. La afectación de estos
sistemas puede disminuir la transcripción genética para la síntesis de
mRNA codificador de proteínas de transporte [17]. Este efecto ha sido
estudiado en modelos animales, observándose una inhibición del flujo y la
secreción biliar al administrar tanto endotoxina [18], como citokinas [19,
20]. Estos resultados indican que la endotoxina, junto con la liberación
de mediadores ocasionada por ella, afecta gravemente el transporte de
todos los compuestos órgánicos biliares, mediante un defecto de síntesis.
La endotoxemia y las citokinas proinflamatorias también producen una
inhibición de la secreción de aniones inorgánicos como el bicarbonato
(Figura 1).
3.2 A nivel celular
La
endotoxemia y el aumento de citokinas originan una mayor expresión de
moléculas endoteliales de adhesión, como la ICAM-1, e integrinas como la
Mac-1. Estas moléculas, originan interacciones celulares y finalmente
migración de neutrófilos hacia el espacio pericelular con liberación de
enzimas (elastasas y proteasas) y radicales libres. Se provoca así la
lesión celular, tanto a nivel sinusoidal como canalicular. A este proceso
pueden contribuir aunque en menor medida, la inducción de la síntesis de
NO [21, 22] y la liberación de noradrenalina intestinal [23, 24]. También
se origina inflamación portal, y desaparición de las proteínas de conexión
entre hepatocitos (conexinas), y contráctiles del canalículo (Figura 2) lo
que conduce a un mayor estasis biliar.
Figura 2
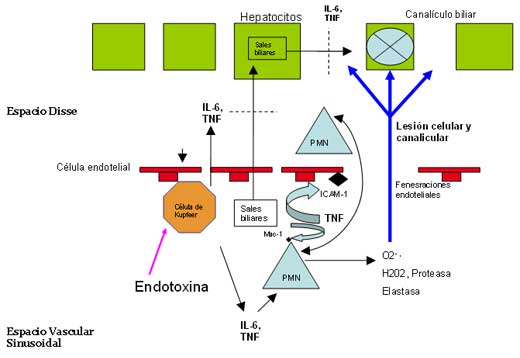
A nivel histológico puede observarse
hiperplasia de las células de Kupffer, infiltrados portales por
mononucleares, y obstrucción de los sinusoides hepáticos por agregados
celulares, disminuyendo el flujo sanguíneo y el área vascular efectivos.
La isquemia, y sobre todo la lesión celular por mediadores y neutrófilos,
originan en el canalículo biliar desaparición de las vellosidades,
afectación de su mecanismo contráctil y ocupación de su interior con
material celular [25]. Todos estos fenómenos conducen a hipoperfusión,
exfoliación endotelial, migración de neutrófilos y finalmente apoptosis de
hepatocitos circundantes y esteatosis [13].
El
resultado final de estas alteraciones bioquímicas y estructurales se
traduce en un impedimento a la secreción y transporte de bilirrubina,
ocasionando el proceso clínico de colestasis intrahepática. El hecho
clínico más destacado es la elevación de bilirrubina, fenómeno que ocurre
hasta en un 54% de los episodios de sepsis; hasta en un 34% la cifra de
bilirrubina es mayor de 2 mg/dl [26]. La hiperbilirrubinemia ocurre de
manera desproporcionada al incremento de otras enzimas como fosfatasa
alcalina, GOT y GPT [27]. Obviamente, la elevación de bilurribina es
superior en enfermos con hepatopatía o neoplasia previas y suele preceder
en estos casos a la aparición de cultivos positivos [26]. También es
posible ver hiperlactacidemia por déficit de extracción [28]. Los
trastornos de la coagulación merecen un comentario aparte.
El hígado, no solo produce
mediadores locales, sino que también modifica su síntesis por efecto de
las citokinas para sintetizar factores procoagulantes (PAI, fibrinógeno,
PAF, trombomodulina, etc.) y disminuir los anticoagulantes (plaquetas, AT
III, proteínas C y S, etc.). Se origina así un estado protrombótico que
contribuye importantemente a la afectación capilar [8]. En este hecho se
basa uno de los principales efectos beneficioso de la proteína C activada
[29].
Existe
controversia sobre cómo medir la disfunción hepática. Un método consiste
en el aclaramiento de sustancias como el verde de indocianina o el MEGX.
Kimura y col. [30] en un trabajo sobre 29 enfermos sépticos encontró que
la constante de eliminación del colorante verde de indocianina
discriminaba bien entre supervivientes y fallecidos. Un fallo en el
incremento de aclaramiento a las 120 horas, o bien un valor extremadamente
bajo a las 24 horas de ingreso, constituía un signo de mal pronóstico. Sin
embargo, para otros autores [31], puede haber interferencias de este
método con la presencia de hepatopatía, su extrapolación al flujo hepático
es problemática, y la sensibilidad insuficiente del MEGX en el enfermo
crítico [2].
Sin
embargo, sistemas de medición por pulsioximetría como el Liver Monitor (Pulsion
Medical Systems) parecen prometedores al ofrecer una técnica fiable de
monitorización a pie de cama. Este dispositivo mide el aclaramiento del
verde de indocianina, previa inyección del colorante por una vía central,
mediante un sistema similar a la pulsioximetría, [32, 33]. Los datos
extraídos de este monitor se correlacionan en enfermos críticos con la
supervivencia, incluso de manera similar al SAPS y el APACHE II [34].
El
diagnóstico diferencial de la hiperbilirrubinemia es amplio. Habitualmente
en un paciente crítico habría que considerar diversas etiologías como
colecistitis acalculosa, litiasis biliar, hepatitis farmacológica,
hemólisis y efecto de la nutrición parenteral total (NPT). Por ello, es
preciso realizar pruebas de imagen (ecografía, TAC, pancreatografía por
RMN) no siendo habitualmente necesaria la biopsia hepática para llegar a
un diagnóstico [2], ya que una ecografía que muestra ausencia de
dilatación de vía biliar extrahepática, y la presencia de signos de sepsis
suele ser suficiente.
Sin embargo, la aparición clínica
puede ser tardía con respecto al inicio de la sepsis. En este sentido Wang
y col. [11] han demostrado mediante técnicas colorimétricas en un modelo
murino, que la disfunción celular ocurre antes del establecimiento del
síndrome de respuesta inflamatoria, indicando que el daño hepático
prioritario en la sepsis es de naturaleza autoinmune como ya se ha
comentado.
En líneas
generales no difiere del manejo general de la sepsis. Es necesario un buen
control del proceso inicial erradicando el foco, administrando
antibioterapia adecuada y ofreciendo un buen soporte de los órganos que
fracasan. No obstante los mismos autores dudan del beneficio de una
adecuada resucitación con fluidos en la reversión de la disfunción
hepatocelular [10]. En este sentido, una adecuada elección de los
inotropos puede ser importante. La dobutamina se ha demostrado como un
agente pernicioso en modelos animales, aunque falta su confirmación en
clínica humana [35]. También cuidar de aspectos como una adecuada
oxigenación y nutrición enteral puede ser de ayuda.
En cuanto
a las terapias de soporte específico, todavía se encuentran en una etapa
de experiencia incipiente. Existen en el mercado diferentes soluciones
para el soporte hepático, todas ellas con ventajas e inconvenientes. En
primer lugar, sistemas celulares con función detoxificadora y de síntesis
en fase de ensayo clínico, como el ELAD y el HepatAssist. Estos sistemas
tienen el inconveniente de su dudosa compatibilidad biológica y su elevado
coste de mantenimiento [36].
Los
sistemas acelulares son principalmente depuradores, y tienen la ventaja de
ser más prácticos, ya que su funcionamiento es similar a los sistemas
extracorpóreos utilizados habitualmente en UCI. Entre ellos, el único
sistema aprobado en Europa es el sistema MARS (Molecular Adsorbents
Recirculating System), desarrollado en Alemania en los años noventa por
Stange y Mitzner [37]. Este sistema se basa en una diálisis con albúmina,
dado que esta proteína es el principal transportador plasmático de
sustancias como bilirrubina, ácidos biliares, cobre y aminoácidos
aromáticos (Figura 3).
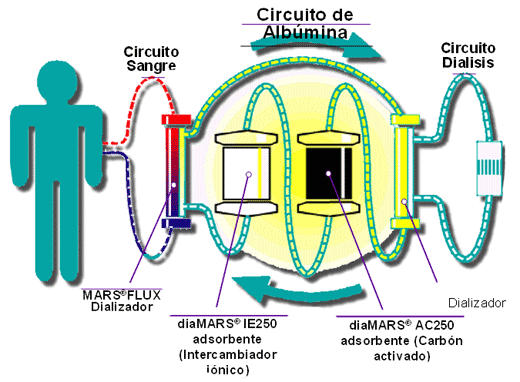
Figura 3
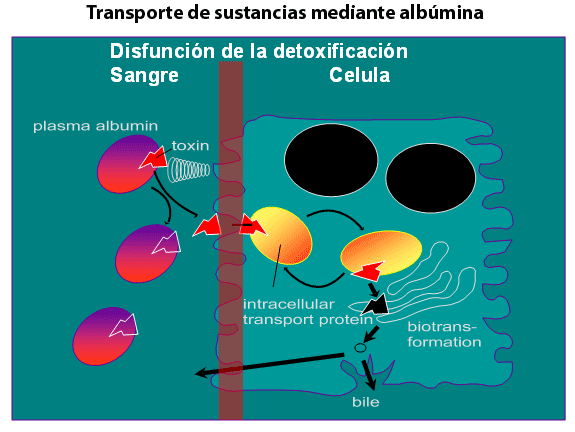
El
líquido dializador es albúmina humana al 20% que es puesta en contacto con
la sangre a través de un cartucho dializador con una membrana de
polisulfona de poro grande (Figura 4). La sangre se depura mediante
extracción de solvente con moléculas pequeñas (amonio, urea, creatinina,
etc.) y transferencia de proteínas grandes a la albúmina. Posteriormente
el solvente se depura en un hemodializador convencional y la albúmina se
regenera para su reutilización mediante un intercambiador de resina y un
intercambiador aniónico [38]. (Figura 5)
Figura 4
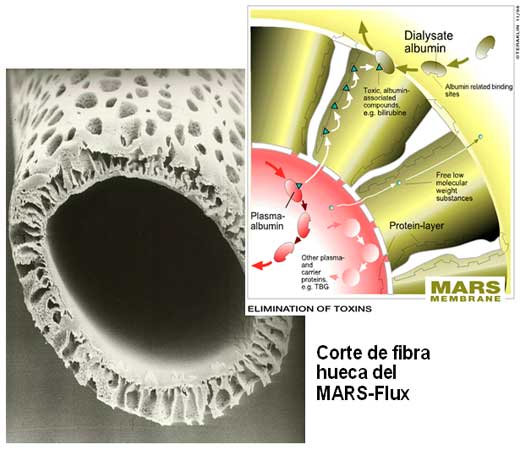
Figura 5
El primer
interrogante para su uso es la ausencia de estudios que nos permitan saber
cuando comenzar a tratar. Los objetivos de la terapia serían la
consecución de estabilidad hemodinámica, la mejoría de la función hepática
y la prevención del desarrollo de fracaso multiorgánico. Guiarse por la
cifra de bilirrubina ha sido la solución más común, tal y como se
desprende de las definiciones de disfunción hepática tomadas de los
principales sistemas pronósticos y de isogravedad. Algunos trabajos
afirman que una bilirrubina superior a 18 mg/dl predice con una alta
sensibilidad y especificidad el fallecimiento; por encima de 24 mg/dl
ningún paciente sobrevive [39]. Por otro lado, hay que tomar en
consideración que la hiperbilirrubinemia es un marcador inespecífico de
disfunción hepática, no cubre todo el amplio espectro de alteraciones
posibles y puede no diferenciar una alteración aguda de una crónica ya
existente [40].
En un
primer momento se ha intentado ser conservador, hasta alcanzar una cifra
de 10 mg/dl. Peek y col. observaron en pacientes con ECMO una
supervivencia del 40% si se trataba con depuración hepática a pacientes
con bilirrubina superior a 18 mg/dl [39]. Sin embargo, la experiencia
enseña que este dintel por sí mismo ya constituye un marcador de elevada
mortalidad, y que los pacientes en fracaso multiorgánico parecen
beneficiarse poco en la práctica diaria tal y como reflejan los resultados
de Wilmer y col. [41]. Una aproximación más fundamentada se basa en el
cociente molar entre la concentración de bilirrubina y albúmina. Cuando
este cociente es superior a ocho [42] la extracción de bilirrubina es más
eficiente. Así, para una albúmina de 2 mg/dl la bilirrubina debería ser 16
mg/dl. En enfermos sépticos la cifra de albúmina suele ser baja, por lo
que el tratamiento podría iniciarse en torno a 10-12 mg/dl de bilirrubina.
Esta cifra podría considerarse como una aproximación razonable, siempre y
cuando existan expectativas de vida fundamentadas en la aplicación
prudente de parámetros clínicos y de índices pronósticos.
Otra
aproximación terapéutica hipotética, teniendo en cuenta la tardía
aparición de hiperbilirrubinemia, sería eliminar mediadores precozmente.
En esta línea, un trabajo de Guo y col. [43] en 24 pacientes con
disfunción multiorgánica y fracaso hepático grave muestra que el sistema
MARS elimina de manera precoz citokinas (TNFα, IL-6, IL-8) y NO, con
disminución del índice SOFA y mejoría clínica; la mortalidad global fue
del 37%, pudiendo transplantarse nueve enfermos. Sin embargo, estos
resultados son preliminares careciéndose de evidencia sólida en las
indicaciones. Otro aspecto a considerar es el elevado coste fuera de
ensayos multicéntricos aleatorizados. Por ello la sepsis constituye la
principal asignatura pendiente para los sistemas de soporte hepático.
Como
regla práctica, aunque cada paciente es debidamente individualizado, en
nuestro centro el soporte hepático mediante MARS en la sepsis se atiene al
siguiente esquema:
-
Soporte
ventilatorio, hemodinámico y renal según criterios estándar
-
Antibioterapia
-
Proteína C
activada de acuerdo a protocolo
-
Control
quirúrgico del foco si procede
-
MARS:
-
Indicación absoluta: Espera de transplante en paciente con hepatopatía
previa.
-
Indicaciones relativas. Dos de las siguientes
-
Bilirrubina total > 10
-
INR > 1,5
o TPT > 40%
-
Encefalopatía grado II o superior
-
GOT ó GPT
> 1.500 UI/L
-
Monitorizar: Mejoría de la función hepática mediante seguimiento de
bilirrubina total, encefalopatía hepática, INR y transaminasas
-
Protocolo
de tratamiento:
-
Intermitente durante 8 horas, salvo fracaso renal anúrico
-
Discontinuar si se cumple el objetivo
-
Continuar
si existe inestabilidad hemodinámica o necesidad de depuración
extrarrenal
|
7. El enfermo cirrótico con sepsis |
Capítulo
aparte merece el enfermo hepático, habitualmente cirrótico, que ingresa en
UCI con deterioro agudo de la función hepática por una infección. En
algunos estudios la sepsis constituye, tras el sangrado gastrointestinal,
la segunda causa de ingreso de estos enfermos [40, 44].
El
pronóstico de los enfermos hepáticos sépticos suele ser ominoso. Antes de
su ingreso es conveniente valorar su estado previo mediante índices
específicos. En este punto, si el intensivista no tiene la ayuda de un
hepatólogo, puede recurrir a índices específicos de fácil uso a pie de
cama. El índice MELD [45], que valora bilirrubina, INR y creatinina
discrimina bien el estadio de enfermedad hepática previa del enfermo:
MELD: 3,8 x ln BT (mg/dl)
+ 11,2 x ln INR + 9,6 x ln creatinina (mg/dl) + 6,4 x etiología (0 si
colestasis o hepatopatía alcohólica; 1 el resto)
Puntuaciones de este índice entre 30 y 39 conllevan una mortalidad del 83%
a los tres meses; puntuaciones iguales o superiores a 40 tienen mortalidad
del 100% en el mismo período.
Una vez
que el enfermo ha ingresado en UCI su mortalidad puede oscilar en torno al
40% según estudios recientes [44], estando la mortalidad hospitalaria en
torno al 45% [40]. Al enfermo cirrótico en UCI se le han aplicado
diferentes sistemas pronósticos. Una puntuación en el APACHE III superior
a 80 puntos indicaba una alta precisión en la estimación de mortalidad
[46]. Aggarwal y col. estiman que la mortalidad del enfermo cirrótico en
UCI se relaciona con el APACHE III, la ventilación mecánica y el uso de
presores, en este orden [44].
El
sistema SOFA de disfunción multiorgánica discriminó en una muestra de 193
pacientes una mortalidad superior al 88% si los enfermos tenían al ingreso
8 o más puntos, e inferior al 4% si la puntuación era menor [40]. Los
valores predictivos positivos y las áreas bajo la curva ROC mostraron una
mayor precisión pronóstica de este índice que el APACHE II y el clásico
Child-Pugh.
Zauner y
col. han diseñado un modelo, el llamado ICCO score, que calcula la
mortalidad al ingreso según la siguiente fórmula:
0,3707 +
(0,0773 x bilirrubina (mg/dl)) - (0,00849 x colesterol (mg/dl)) -(0,0155 x
aclaramiento de creatinina (ml/min)) + (0,1351 x lactato (mmol/l).
La
discriminación diagnóstica de este test es similar al SOFA, con un área
bajo la curva de 0,9. Todos los pacientes con una puntuación mayor de 2,6
murieron [47].
Específicamente, la incidencia de sepsis en pacientes cirróticos
ingresados en UCI es de un 30%, siendo la peritonitis bacteriana la causa
más frecuente de infección [48]. La mortalidad puede alcanzar en estos
casos el 60% [26], relacionándose fundamentalmente con la aparición de
tres de los componentes del SRIS [49], insuficiencia renal e infiltrados
pulmonares [48].
En este
tipo de pacientes la sepsis puede ser un proceso larvado, de difícil
diagnóstico inicial, dado que la propia hepatopatía puede originar
trombocitopenia, hiperfibrinolisis, déficit de síntesis de factores y
coagulación intravascular diseminada. Para dificultar más el diagnóstico
hasta un tercio de los enfermos no presenta signos de respuesta
inflamatoria sistémica [49]. El diagnóstico diferencial puede requerir
métodos auxiliares de imagen y bioquímicos como la procalcitonina. Este
marcador de origen hepático puede diferenciar a pacientes cirróticos
descompensados por infección, de aquellos no infectados. Valores
superiores a 0,58 ng/ml son capaces de discriminar bien entre ambos grupos
de pacientes, con una sensibilidad del 92% y una especificidad del 72%,
siendo superior a parámetros como la Proteína C reactiva o la IL-6 [50].
El
tratamiento de estos enfermos no difiere esencialmente de cualquier otro
enfermo séptico, excepto en la necesidad de soporte de la función
hepática, con fármacos en el caso de la encefalopatía, diálisis para el
síndrome hepatorrenal, o de una manera más global, con el sistema MARS.
Con este sistema de depuración disminuye el grado de encefalopatía [51-53]
y la disfunción renal de estos enfermos [54-56]. Los únicos estudios que
han evaluado la aplicación de este sistema sobre la mortalidad demuestran
una reducción de la misma en enfermos en espera de transplante [57] y en
enfermos con síndrome hepatorrenal [58].
El
estudio con mayor grado de evidencia, realizado por Heemann y col. sobre
una serie de 20 enfermos con hepatopatía crónica reagudizada, mostró que
los enfermos con bilirrubina superior a 20 mg/dl tratados con MARS
experimentaban una mejoría de los parámetros bioquímicos y una reducción
significativa de la mortalidad [59]. Sin embargo, sólo tres de estos
enfermos tuvieron un diagnóstico de infección, aunque en estos hubo una
clara mejoría. Asimismo, la existencia de diferencias en variables
clínicas de interés entre ambos grupos, y la baja muestra del estudio por
una interrupción precoz del comité ético, son factores que hacen necesario
esperar, en opinión de expertos [60], a dos estudios multicéntricos en
marcha.
En
nuestro centro, y basados en protocolos europeos y nacionales, hemos
establecido una serie de normas para el ingreso de estos enfermos, y su
tratamiento, si procede, con sistema MARS:
|
Tabla:
Crierios para el uso de MARS en pacientes sépticos |
| |
| 1.-
Indicaciones genéricas |
Enfermos entre 18 y 65
años (límite al transplante)
Enfermos incluidos en la lista de trasplante activa |
| 2.-
Contraindicaciones absolutas |
| Escala MELD
≥
40 puntos |
| 3.-
Contraindicaciones relativas |
Previamente a realizar
tratamientos electivos con MARS si:
- No candidato a transplante:
Edad superior a 65 años
hepatocarcinoma u otra
neoplasia maligna
Cardiopatía avanzada
EPOC con oxígeno
domiciliario
Otra enfermedad sistémica
grave
Trombosis portal
Alcoholismo activo
8valorar individualmente)
- Valorar puntuación ICCO > 2,6 en el primer día de ingreso en UCI
(mortalidad 100%; también útil como criterio electivo de ingreso)
- Signos de CID
- Sangrado activo: necesidad de transfusión e inestabilidad
hemodinámica
- Shock séptico sin respuesta a antibióticos
- Funguemia
- Hemorragia aguda sin respuesta a tratamiento estándar
|
Manejo
del enfermo con MARS:
-
Monitorizar: Mejoría de la función hepática mediante seguimiento de
Bilirrubina total, encefalopatía hepática, INR y transaminasas
-
Protocolo de
tratamiento:
-
Intermitente durante 8 horas, salvo fracaso renal anúrico
-
Discontinuar si se cumple el objetivo
-
Continuar
si existe inestabilidad hemodinámica o necesidad de depuración
extrarrenal.
|
-
Vincent JL, Moreno R, Takala J, Willatts S, De
Mendonca A, Bruining H et al. The SOFA
(Sepsis-related Organ Failure Assessment) score to describe organ
dysfunction/failure. On behalf of the Working Group on Sepsis-Related
Problems of the European Society of Intensive Care Medicine. Intensive
Care Med 1996; 22(7):707-710.PM:8844239
-
Lopez Alvarez JM, Sancho RH, De Irala EJ, De la Mata
GM, Robles Arista JC, Fernandez-Crehuet R. The monoethil-glicinexilidide
formation test (MEGX-test) in the assessment of liver dysfunction in
critically ill patients. [Spanish]. Medicina Intensiva 1998; Vol 22:
365-372)
-
Fuchs S, Bogomolski-Yahalom V, Paltiel O, Ackerman Z.
Ischemic hepatitis: clinical and laboratory observations of 34 patients.
J Clin Gastroenterol 1998; 26(3):183-186. PM:9600366
-
Sikuler E, Guetta V, Keynan A, Neumann L, Schlaeffer
F. Abnormalities in bilirubin and liver enzyme levels in adult patients
with bacteremia. A prospective study. Arch Intern Med 1989;
149(10):2246-2248. PM:2802891
-
Trauner M, Fickert P, Stauber RE.
Inflammation-induced cholestasis. J Gastroenterol Hepatol 1999;
14(10):946-959. PM:10530489
-
Russell JA, Singer J, Bernard GR, Wheeler A,
Fulkerson W, Hudson L et al. Changing pattern of organ dysfunction in
early human sepsis is related to mortality. Crit Care Med 2000;
28(10):3405-3411. PM:11057793
-
Marrero J, Martinez FJ, Hyzy R. Advances in critical
care hepatology. Am J Respir Crit Care Med 2003; 168(12):1421-1426.
PM:14668256
-
Szabo G, Romics L, Jr., Frendl G. Liver in sepsis and
systemic inflammatory response syndrome. Clin Liver Dis 2002;
6(4):1045-1066. PM:12516206
-
Seeto RK, Fenn B, Rockey DC. Ischemic hepatitis:
clinical presentation and pathogenesis. Am J Med 2000; 109(2):109-113.
PM:10967151
-
Wang P, Ba ZF, Ayala A, Chaudry IH. Hepatocellular
dysfunction persists during early sepsis despite increased volume of
crystalloid resuscitation. J Trauma 1992; 32(3):389-396. PM:1548729
-
Wang P, Ba ZF, Chaudry IH. Hepatocellular dysfunction
occurs earlier than the onset of hyperdynamic circulation during sepsis.
Shock 1995; 3(1):21-26. PM:7850575
-
Crawford JM, Boyer JL. Clinicopathology conferences:
inflammation-induced cholestasis. Hepatology 1998; 28(1):253-260.
-
Moseley RH. Sepsis and cholestasis. Clin Liver Dis
2004; 8(1):83-94. PM:15062195
-
Vos TA, Hooiveld GJ, Koning H, Childs S, Meijer DK,
Moshage H et al. Up-regulation of the multidrug resistance genes, Mrp1
and Mdr1b, and down-regulation of the organic anion transporter, Mrp2,
and the bile salt transporter, Spgp, in endotoxemic rat liver.
Hepatology 1998; 28(6):1637-1644. PM:9828229
-
Byrne JA, Strautnieks SS, Mieli-Vergani G, Higgins
CF, Linton KJ, Thompson RJ. The human bile salt export pump:
characterization of substrate specificity and identification of
inhibitors. Gastroenterology 2002; 123(5):1649-1658. PM:12404239
-
Buchler M, Konig J, Brom M, Kartenbeck J, Spring H,
Horie T et al. cDNA cloning of the hepatocyte canalicular isoform of the
multidrug resistance protein, cMrp, reveals a novel conjugate export
pump deficient in hyperbilirubinemic mutant rats. J Biol Chem 1996;
271(25):15091-15098. PM:8662992
-
Wang B, Cai SR, Gao C, Sladek FM, Ponder KP.
Lipopolysaccharide results in a marked decrease in hepatocyte nuclear
factor 4 alpha in rat liver. Hepatology 2001; 34(5):979-989. PM:11679969
-
Roelofsen H, Schoemaker B, Bakker C, Ottenhoff R,
Jansen PL, Elferink RP. Impaired hepatocanalicular organic anion
transport in endotoxemic rats. Am J Physiol 1995; 269(3 Pt 1):G427-G434.
PM:7573454
-
Wang P, Ba ZF, Chaudry IH. Mechanism of
hepatocellular dysfunction during early sepsis. Key role of increased
gene expression and release of proinflammatory cytokines tumor necrosis
factor and interleukin-6. Arch Surg 1997; 132(4):364-369. PM:9108756
-
Whiting JF, Green RM, Rosenbluth AB, Gollan JL. Tumor
necrosis factor-alpha decreases hepatocyte bile salt uptake and mediates
endotoxin-induced cholestasis. Hepatology 1995; 22(4 Pt 1):1273-1278.
PM:7557881
-
Liaudet L, Soriano FG, Szabo C. Biology of nitric
oxide signaling. Crit Care Med 2000; 28(4 Suppl):N37-N52. PM:10807315
-
Shieh P, Zhou M, Ornan DA, Chaudry IH, Wang P.
Upregulation of inducible nitric oxide synthase and nitric oxide occurs
later than the onset of the hyperdynamic response during sepsis. Shock
2000; 13(4):325-329. PM:10774623
-
Yang S, Koo DJ, Zhou M, Chaudry IH, Wang P. Gut-derived norepinephrine
plays a critical role in producing hepatocellular dysfunction during
early sepsis. Am J Physiol Gastrointest Liver Physiol 2000;
279(6):G1274-G1281.PM:11093951
-
Yang S, Zhou M, Chaudry IH, Wang P. Norepinephrine-induced
hepatocellular dysfunction in early sepsis is mediated by activation of
alpha2-adrenoceptors. Am J Physiol Gastrointest Liver Physiol 2001;
281(4):G1014-G1021. PM:11557522
-
Hirata K, Ikeda S, Honma T, Mitaka T, Furuhata T,
Katsuramaki T et al. Sepsis and cholestasis: basic findings in the
sinusoid and bile canaliculus. J Hepatobiliary Pancreat Surg 2001;
8(1):20-26. PM:11294287
-
Franson TR, Hierholzer WJ, Jr., LaBrecque DR.
Frequency and characteristics of hyperbilirubinemia associated with
bacteremia. Rev Infect Dis 1985; 7(1):1-9. PM:3983523
-
Moseley RH. Sepsis and cholestasis. Clin Liver Dis
1999; 3(3):465-475. PM:11291234
-
Chrusch C, Bands C, Bose D, Li X, Jacobs H, Duke K et
al. Impaired hepatic extraction and increased splanchnic production
contribute to lactic acidosis in canine sepsis. Am J Respir Crit Care
Med 2000; 161(2 Pt 1):517-526. PM:10673195
-
Bernard GR, Vincent JL, Laterre PF, LaRosa SP,
Dhainaut JF, Lopez-Rodriguez A et al. Efficacy and safety of recombinant
human activated protein C for severe sepsis. N Engl J Med 2001;
344(10):699-709. PM:11236773
-
Kimura S, Yoshioka T, Shibuya M, Sakano T, Tanaka R,
Matsuyama S. Indocyanine green elimination rate detects hepatocellular
dysfunction early in septic shock and correlates with survival. Crit
Care Med 2001; 29(6):1159-1163. PM:11395594
-
Smets D, Spapen H, Diltoer M, Nguyen DN, Hubloue I,
Huyghens L. Liver perfusion and hepatocellular inflammatory response in
sepsis. Acta Clin Belg 1999; 54(4):201-206. PM:10544510
-
Sakka SG, Reinhart K, Wegscheider K, Meier-Hellmann
A. Comparison of cardiac output and circulatory blood volumes by
transpulmonary thermo-dye dilution and transcutaneous indocyanine green
measurement in critically ill patients. Chest 2002; 121(2):559-565.
PM:11834672
-
Sakka SG, Reinhart K, Meier-Hellmann A. Comparison of
invasive and noninvasive measurements of indocyanine green plasma
disappearance rate in critically ill patients with mechanical
ventilation and stable hemodynamics. Intensive Care Med 2000;
26(10):1553-1556. PM:11126271
-
Sakka SG, Reinhart K, Meier-Hellmann A. Prognostic
value of the indocyanine green plasma disappearance rate in critically
ill patients. Chest 2002; 122(5):1715-1720. PM:12426276
-
Tighe D, Moss R, Heywood G, al Saady N, Webb A,
Bennett D. Goal-directed therapy with dopexamine, dobutamine, and volume
expansion: effects of systemic oxygen transport on hepatic
ultrastructure in porcine sepsis. Crit Care Med 1995; 23(12):1997-2007.
PM:7497722
-
Demetriou AA, Rozga J, Podesta L, Lepage E, Morsiani
E, Moscioni AD et al. Early clinical experience with a hybrid
bioartificial liver. Scand J Gastroenterol Suppl 1995; 208:111-117.
PM:7777790
-
Stange J, Mitzner SR, Risler T, Erley CM, Lauchart W,
Goehl H et al. Molecular adsorbent recycling system (MARS): clinical
results of a new membrane-based blood purification system for
bioartificial liver support. Artif Organs 1999; 23(4):319-330.
PM:10226696
-
Herrera Gutierrez ME, Seller G, oz A, Lebron M,
Aragon C. Extracorporeal liver support: Current situation and future
prospects. [Spanish]. Medicina Intensiva, 2004;Vol 28(4): 211-218.
-
Peek GJ, Killer HM, Sosnowski MA, Firmin RK. Modular
extracorporeal life support for multiorgan failure patients. Liver 2002;
22 Suppl 2:69-71. PM:12220309
-
Wehler M, Kokoska J, Reulbach U, Hahn EG, Strauss R.
Short-term prognosis in critically ill patients with cirrhosis assessed
by prognostic scoring systems. Hepatology 2001; 34(2):255-261.
PM:11481609
-
Wilmer A, Nevens F, Evenepoel P, Hermans G, Fevery J.
The Molecular Adsorbent Recirculating System in patients with severe
liver failure: clinical results at the K.U. Leuven. Liver 2002; 22 Suppl
2:52-55. PM:12220305
-
Lee KH, Wendon J, Lee M, Da Costa M, Lim SG, Tan KC.
Predicting the decrease of conjugated bilirubin with extracorporeal
albumin dialysis MARS using the predialysis molar ratio of conjugated
bilirubin to albumin. Liver Transpl 2002; 8(7):591-593. PM:12089711
-
Guo LM, Liu JY, Xu DZ, Li BS, Han H, Wang LH et al.
Application of Molecular Adsorbents Recirculating System to remove NO
and cytokines in severe liver failure patients with multiple organ
dysfunction syndrome. Liver Int 2003; 23 Suppl 3:16-20. PM:12950956
-
Aggarwal A, Ong JP, Younossi ZM, Nelson DR,
Hoffman-Hogg L, Arroliga AC. Predictors of mortality and resource
utilization in cirrhotic patients admitted to the medical ICU. Chest
2001; 119(5):1489-1497. PM:11348958
-
Kamath PS, Wiesner RH, Malinchoc M, Kremers W,
Therneau TM, Kosberg CL et al. A model to predict survival in patients
with end-stage liver disease. Hepatology 2001; 33(2):464-470.
PM:11172350
-
Zimmerman JE, Wagner DP, Seneff MG, Becker RB, Sun X,
Knaus WA. Intensive care unit admissions with cirrhosis:
risk-stratifying patient groups and predicting individual survival.
Hepatology 1996; 23(6):1393-1401. PM:8675156
-
Zauner CA, Apsner RC, Kranz A, Kramer L, Madl C,
Schneider B et al. Outcome prediction for patients with cirrhosis of the
liver in a medical ICU: a comparison of the APACHE scores and
liver-specific scoringsystems. Intensive Care Med 1996; 22(6):559-563.
PM:8814471
-
Singh N, Gayowski T, Wagener MM, Marino IR. Outcome
of patients with cirrhosis requiring intensive care unit support:
prospective assessment of predictors of mortality. J Gastroenterol 1998;
33(1):73-79. PM:9497225
-
Rolando N, Wade J, Davalos M, Wendon J, Philpott-Howard
J, Williams R. The systemic inflammatory response syndrome in acute
liver failure. Hepatology 2000; 32(4 Pt 1):734-739. PM:11003617
-
Connert S, Stremmel W, Elsing C. Procalcitonin is a
valid marker of infection in decompensated cirrhosis. Z Gastroenterol
2003; 41(2):165-170. PM:12592597
-
Ash SR. Extracorporeal blood detoxification by
sorbents in treatment of hepatic encephalopathy. Adv Ren Replace Ther
2002; 9(1):3-18. PM:11927902
-
Aviles J, Macia M, Morales S, Perez F, Moreno A,
Navarro J et al. [Efficiency of dialysis with albumin in the treatment
of patients with advanced hepatic insufficiency: initial experience with
the MARS system in Spain]. Nefrologia 2001; 21(4):376-385. PM:11816514
-
Mullhaupt B, Kullak-Ublick GA, Ambuhl P, Maggiorini
M, Stocker R, Kadry Z et al. First clinical experience with Molecular
Adsorbent Recirculating System (MARS) in six patients with severe acute
on chronic liver failure. Liver 2002; 22 Suppl 2:59-62. PM:12220307
-
Mitzner SR, Stange J, Klammt S, Risler T, Erley CM,
Bader BD et al. Improvement of hepatorenal syndrome with extracorporeal
albumin dialysis MARS: results of a prospective, randomized, controlled
clinical trial. Liver Transpl 2000; 6(3):277-286. PM:10827226
-
Mitzner SR, Klammt S, Peszynski P, Hickstein H,
Korten G, Stange J et al. Improvement of multiple organ functions in
hepatorenal syndrome during albumin dialysis with the molecular
adsorbent recirculating system. Ther Apher 2001; 5(5):417-422.
PM:11778928
-
Mullen KD. Treatment of hepatorenal syndrome: lessons from the MARS
trial. Hepatology 35[2], 492-493. 2002.
-
Stange J. New extracorporeal liver support for chronic liver disease
complicated by cholestasis - results of a prospective controlled
randomized two center trial. Mitzner S, Klammt S, Loock J, Treichel U,
Gerken G, Schmidt R et al., editors. J Hepatology SI, 45 A 1289. 2001.
-
Mitzner S, Loock J, Peszynski P, Klammt S,
Majcher-Peszynska J, Gramowski A et al. Improvement in central nervous
system functions during treatment of liver failure with albumin dialysis
MARS--a review of clinical, biochemical, and electrophysiological data.
Metab Brain Dis 2002; 17(4):463-475. PM:12602522
-
Heemann U, Treichel U, Loock J, Philipp T, Gerken G,
Malago M et al. Albumin dialysis in cirrhosis with superimposed acute
liver injury: a prospective, controlled study. Hepatology 2002; 36(4 Pt
1):949-958. PM:12297843
-
Kamath PS. Is there life in MARS?
Hepatology 2002; 36(4 Pt 1):1017-1019. PM:12297854
Vicente Gómez Tello
Clínica Moncloa, Madrid
©REMI,
http://remi.uninet.edu. Diciembre 2004.
Palabras clave:
Sepsis, Sepsis grave, Disfunción hepática, cirrosis hepática, MARS.
Busque en REMI con Google:
Envía tu comentario para su
publicación |
|
|
|



